 |
 |
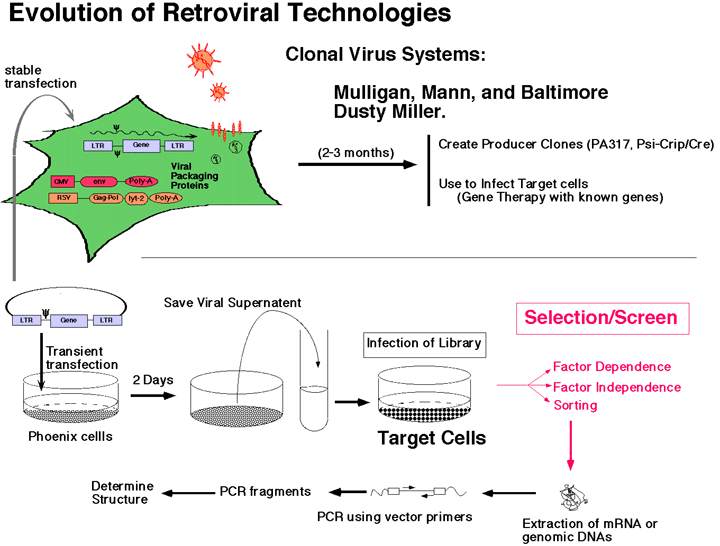
Genetic Screens in Mammalian Cells using Retroviruses
This page discusses some of the theoretical aspects and potential pitfalls of library screening and enrichment for a given phenotype.
Screening for a rare event against a background of unwanted events is one of the great pleasures of genetics. The trick, and much of the creativity in genetics is creating a screening system that distinguishes the event of interest from events that have no interest at the moment of the screen. Essentially, you are on a fishing trip for the unknown and it all depends on what kind of bait you use for your hook.
Such phenotypic screening takes a variety of forms. Modern applications of genetic screening are two-hybrid screens, lambda phage genomic library screens, and complementation cloning in yeast. Extended examples of genetic selection principles applied to other types of "searches" include pharmaceutical searches for drugs against disease or phage display screens for phage that bind to target proteins.
In the Nolan lab we use genetic screens by applying retroviral libraries to mammalian signaling systems. In general, the events for which we are searching/fishing are rare, therefore some thought and consideration must be made as to how to approach such screens.
Before embarking on any phenotypic or genetic screen several factors should be considered:
-
Event Frequency
-
Library Size
-
False Positives
-
Recovery
-
Efficiency
What
is the frequency of the event for which one is searching?
You should make some estimate of what the likelihood or rarity of
the the even is for which you are fishing or searching. This will tell
you how long to look, where to look, how to look, and in how many cells
in a population you must look for the event of interest. There are several
pages following this one and linked herein that will explain many of
these concepts.
Is
the "library" you have in hand which you plan to search large enough
to contain the event for which you are looking?
If the library you plan to screen contains only 10^4 members and the
event is expected at 1 in 10^6 then it is unlikely you will find the
event in your library. However, to be 95% confident you've screened
the entire library, you must search through roughly 3 times the size
of your library, as determined by the following equation:
{ln(1-.95)/ln(1-1/(Library Size))}
What
is the frequency of false positive events (background)? Is the background
event sporadic or heritable?
A "sporadic" background event is pure noise. For instance, let's say
you were sorting cells by Flow Cytometry for a rare event like expression
of a human surface protein after deliveryof a human retroviral cDNA
library to a population of cells. Let's say that the event is expected
in the retroviral cDNA library at 1 in 10^6. Let's say you sorted 1000
event-positive cells after sorting through 10^8 cells in a library.
You grow up the sorted cells. Simple math (let's forget pure statistics
for a moment) would predict that in that library of 10^8 cells there
should have been only 100 positive cells. That means 900 cells are background,
unwanted (presumably) events (the background frequency of false positives
in the original screen can be deduced to have been 9 in 10^6). You have
however, presumably enriched for the desired phenotype event. Therefore,
if the 1000 cells are cultured and expanded, then reanalyzed, one should
observe an enrichment of positive events if the background was sporadic
(not heritable). Now the percentage of positive cell events should approach
10%. For many purposes this would suffice to clone cells or sort with
confidence that you are isolating "real" events.
However, if the background of false positive events is heritable, you
can have some trouble. For instance, let's say you are searching for
a cDNA that activates a given gene's expression (like activating some
cell surface marker (CSM)). Assume that the frequency of the cDNA for
which you are searching is 1 in 10^6. Let's say that epigenetic or random
mutation provides that in a pure population of virgin CSM-negative cells
that 9 cells out of 10^6 spontaneously and without provocation convert
to expression of CSM and that such expression change is heritable. Thus,
if you analyze 1 x 10^8 cells and sort the positive cells you will get
1000 cells, as in the previous paragraph. However, when you REANALYZE
the cells after culturing them, you will find that nearly 100% of the
cells continue to be positive. However, the majority of the cells are
false positive and the minority (10%) are the event for which you search.
How
efficient is the screen at the level of recovery?
That is, even if you isolate a cell that displays the desired event,
what is the chance the cell might die before you can determine the nature
of the event? What is your ability to efficiently transfer the event?
How many rounds of selection or enrichment must you undertake before
the event of interest can be recognized above background?
This depends on a number of factors that have been outlined above. The
equations below outline some of the numeric considerations that must
be applied before any screening project can be undertaken with confidence.
Check out the Excel file I made for downloading below for help with
this.
Of course, you can ignore all this math stuff and just get darn lucky...
Much of the above is encapsulated in a MS Excel spreadsheet on the screening pages for analysis and plugging in of your own numbers. Some explanations can be seen in the first example and second example of screening.
The available download is a MS Excel file which does the appropriate calculations. Apply these equations with caution as they will provide only a best-guess approximation. The Nolan lab assumes no responsibility for wasted graduate student or postdoctoral years spent on screening projects undertaken after application of this equation set. It's your screen and your assumptions you plug in to the equations-- in other words, you're on your own!!
Examples:
-
Our first first example of a screen shows how to graph and understand the limitations of library screening using your own selection systems and assumptions.
-
Our second example of a screen refines the ideas of the first screen.
![]()
Home Page |
Interests | Members
|
MTA Forms | Plasmid
Maps | Retroviral Systems
| Genetic Screens | Library
Systems | Protocols
| Tutorials |
Publications | Contact
| Virus Chat